first published : 27.10.2020
This is a OPINION PIECE and should be interpreted with CAUTION!
The text is ‘overtly simplified’ for general audience and DOES NOT CLAIM scientific accuracy but is only intended to present easy to understand information.
Why the SARS-CoV-2 genome is evolving?
The SARS-CoV-2 genome is nearly 30,000 bases long and undergoes a rapid replication process (‘creating identical copies’) in an infected host (each host cell can produce thousands of viral RNA copies, which in effect would be several billions of viral RNA copies in humans). And then following a transmission to another host, undergoes another round of rapid replication process, and so on. During these replication processes, random ‘mutations’ (errors/mistakes in copying/creating idential genomes) can happen, although the chances are miniscule, only a couple of wrong bases in for every 10-100 million bases copied. Now, even though these chance mutations (naturally occuring) are very small, some of these mutation can happen in genomic regions which has a functional role, like in virus transmission pathways, or viral pathogenesis, or infection mechanisms, or structural stability, and so on. Next, if this pool-of-viruses are happen to randomly infect a group of hosts, one of the evolved/mutated virus genome may have an ‘advantage’ over the other viruses in a sub-set of hosts (as the hosts also differ amongst themselves). In fictional words, the European population (hosts) may be more vulnerable (merely statistically speaking) for a particular ‘mutated virus’ compared to another distinct host population, say the residents of Indian-subcontinent. The environmental factors also contribute but not to digress too much, we keep the discussion to mutations in SARS-CoV-2 genomes. And an epidemic ensues when one infected host trasmits the virus to more than one subsequent host and it continues!
What does this ‘evolving genome’ means in terms of the ongoing pandemic?
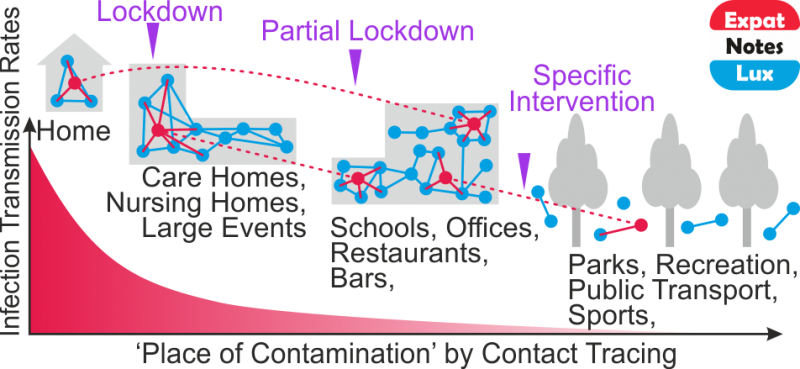
Before we discuss that, let us quickly look at the current pandemic state in Europe (as of Oct 2020). So, each European country recommended and undertook a different set of ‘measures’ in different time periods for different sectors of the society to reduce virus transmission, the population make-up is different, the local environment and weather is different, the population movement is quite ‘limited’ in comparison to Feb-April 2020, but still the whole Europe seems to be affected by the ongoing ‘second wave’, with several countries experiencing higher severity than the ‘first wave’. So, a fundamental question would be – what is the common factor? This is where PROBABLY, one of the common factors could the virus itself, and more specifically a common virus strain (or sets of strains).
Can we ‘visualize’ the evolving mutations in Europe?
Researchers have ‘sequenced‘ the SARS-CoV-2 genome across the world since the first cases reported in Dec 2019 and over 3,400 SARS-CoV-2 genome sequences are available at Nextstrain from European researchers (as of Oct 2020). Below is a snapshot of 425 genome sequences from different laboratories from Belgium, France, Germany and Luxembourg, (circles) while the backgroud (lines) represent the number of ‘mutations’ (y-axis) compared to the first sequenced genome over time (x-axis) for over 3,400 SARS-CoV-2 genomes. Seven major clades (GR, G, GH, O, S, L and V) are shown as well which are like seven sub-groups which are different from each other (starting with a single but important mutation) and then although there are many more mutations in different sites/places in the genomes, the starting ‘single mutation’ which rendered some ‘functional’ difference is intact (and hence a clade). The functional aspects of the clades are not discussed as it not the purpose here, but only to highlight the number of mutations are slowly increasing over time.
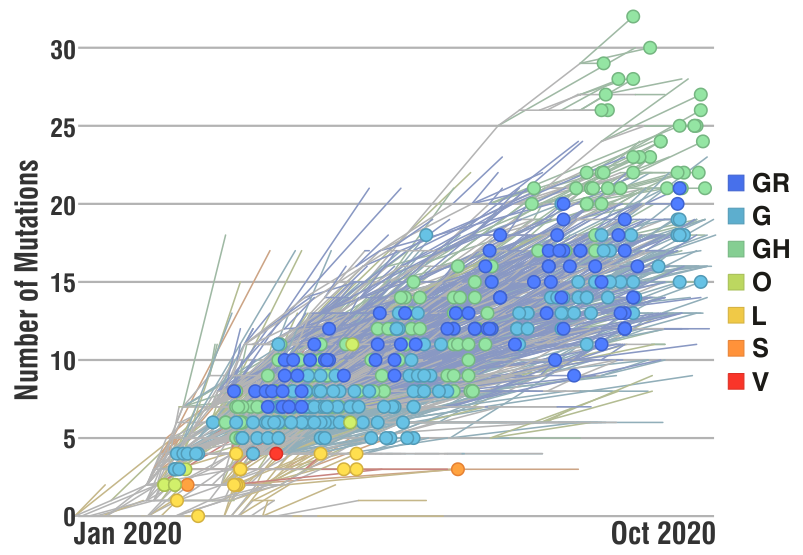
Does these mutations have any ‘real-world’ significance in the current pandemic scenario?
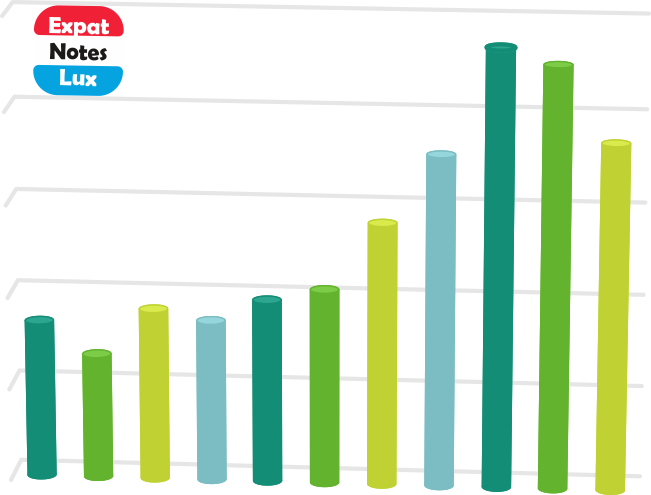
Broadly speaking, these ‘mutations’ (evolving genomes) have two main ‘real-world’ significance. One, it COULD partly explain the ‘re-infections‘ in individuals (as in case of seasonal flu which may infect the same individual in different seasons) and also partly the ongoing ‘second wave’, as these ‘mutated’ genomes although have almost similar pathogenesis, they could have different ‘prevalence’ in the host population at different times. Below is a snapshot of the seven different ‘clades’ and there prevalence in Belgium, France, Germany and Luxembourg (NOTE: all countries did not sequence equal number of genomes over time, and the frequencies are not accurate in its strict sense. The frequency was calculated as 100% with the available number of sequences at each distinct time point.)
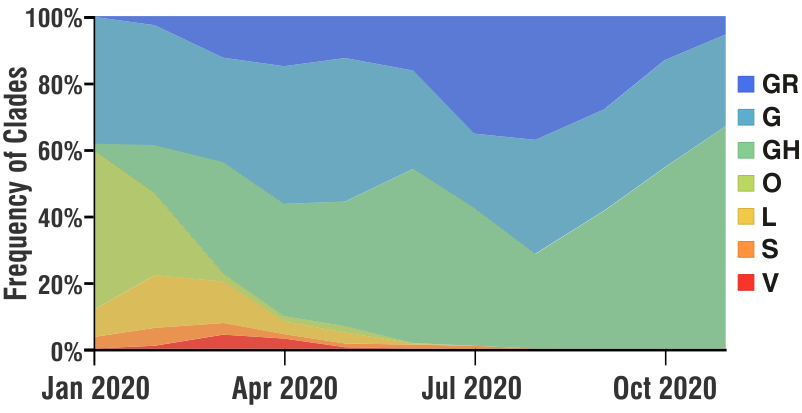
The second ‘real-world’ significance is in vaccine development and effectiveness. For example, if the ‘current vaccine’ works really effectively with the prevailing ‘mixture’ of SARS-CoV-2 genomes, by the time the vaccine is available publicly (in several months at best), it could happen that the SARS-CoV-2 genomes have further evolved / mutated and show a less reactivity towards the vaccine. Having said that, it may be helpful to mention that we would probably need a ‘cocktail’ of vaccine with different targets and different mechanisms to counter the virus. We anticipate that the research and pharmaceutical world would also evolve according to the needs of this globalc pandemic.